
Hypersensitivitätspneumonitis – wie oft denken wir Pathologen nicht daran?
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Die Hypersensitivitätspneumonitis (HP) ist eine immunvermittelte interstitielle Lungenerkrankung, die durch Immunreaktionen auf inhalierte Antigene verursacht wird. Die Diagnose stützt sich auf die Anamnese, klinische Untersuchung, Radiologie, bronchoalveoläre Lavage und, falls erforderlich, eine Lungenbiopsie. Ein zentrales histologisches Merkmal ist das Vorhandensein nichtnekrotisierender Granulome. Allerdings können auch andere pathologische Veränderungen im Lungenparenchym auf eine HP hinweisen, was die Frage aufwirft: Wie oft bleiben diese unbemerkt?
Keypoints
-
Die Hypersensitivitätspneumonitis weist unterschiedliche pathohistologische Veränderungen auf.
-
Sie bleibt oft eine unterdiagnostizierte Erkrankung, häufig aufgrund von Probenahmeproblemen.
-
Das Vorhandensein von Granulomen ist für die Diagnose nicht immer erforderlich.
-
Eine genaue Diagnose ist entscheidend für die Bestimmung der geeigneten therapeutischen Vorgehensweise.
Hypersensitivitätspneumonitis als interstitielle Lungenerkrankung
Interstitielle Lungenerkrankungen (ILD) umfassen eine heterogene Gruppe nichtneoplastischer Lungenerkrankungen, die durch unterschiedliche Grade von Entzündung und Fibrose gekennzeichnet sind. Diese Erkrankungen betreffen primär die interalveolären Septen, können jedoch auch die Lufträume einbeziehen, wobei einige charakteristische morphologische Merkmale wie Granulome aufweisen. Während viele ILD, wie atypische Pneumonien, eine identifizierbare infektiöse Ursache und eine günstige Prognose haben, stehen andere, darunter die Hypersensitivitätspneumonitis (HP) und die Pneumokoniosen, mit Umweltbelastungen in Verbindung.
HP ist eine chronisch-entzündliche und/oder fibrotische Lungenerkrankung, die das Lungenparenchym und die kleinen Atemwege betrifft. Sie wird durch eine immunvermittelte Reaktion auf eingeatmete Antigene ausgelöst, die in der Regel kleiner als 5 Mikrometer sind und die distalen Alveolen erreichen, wo sie eine Entzündungsreaktion hervorrufen. Laut aktuellen Leitlinien wird HP in fibrotische und nicht-fibrotische Formen unterteilt, eine Klassifikation, die sich mit der radiologischen Einteilung deckt und prognostische Bedeutung hat. Die Diagnose von HP bleibt komplex und erfordert einen multidisziplinären Ansatz, der klinische, radiologische und histopathologische Befunde integriert.
Ätiologie und Epidemiologie der HP
HP ist mit über 50 berufsbedingten und umweltbedingten Expositionen assoziiert, darunter Bakterien, Pilze, tierische Proteine und niedermolekulare Chemikalien. Die Ätiologie variiert je nach geografischen und beruflichen Faktoren. In Nordamerika sind Vogelantigene und Schimmelpilze häufige Auslöser, während HP in feuchten Regionen wie Japan und Indien mit schimmelbelasteten Klimaanlagen in Verbindung gebracht wird. HP kann in allen Altersgruppen auftreten und betrifft Männer und Frauen gleichermaßen. Interessanterweise ist die Prävalenz bei Rauchern geringer. Dies wird auf eine verminderte Lymphozytenproliferation und Makrophagenaktivierung zurückgeführt, wodurch die Immunantwort auf eingeatmete Antigene abgeschwächt wird.
Die Identifizierung und Vermeidung des sensibilisierenden Antigens sind für Diagnose und Therapie entscheidend. Dies gestaltet sich jedoch oft schwierig, da in bis zu 60% der Fälle von fibrotischer HP keine spezifische Exposition nachgewiesen werden kann. Diese diagnostische Unsicherheit erfordert eine umfassende klinische Bewertung und einen multidisziplinären Ansatz.
Klinisches Bild der HP
Die klinische Präsentation der HP ist variabel und wird durch die Antigenexposition, das Ausmaß der Fibrose und die individuelle Suszeptibilität beeinflusst. Die Symptome sind weitgehend unspezifisch.
Bei nichtfibrotischer HP treten frühe Symptome wie trockener Husten, Dyspnoe und systemische Beschwerden wie Fieber, Schüttelfrost und Abgeschlagenheit auf, oft in zeitlichem Zusammenhang mit der Antigenexposition. Die Entfernung des auslösenden Antigens kann zur Krankheitsauflösung oder Stabilisierung führen. Wiederholte Exposition kann jedoch zur Krankheitsprogression beitragen.
Die fibrotische HP hingegen zeigt sich durch schleichend zunehmende Belastungsdyspnoe und persistierenden Husten. Im Vergleich zur fibrotischen Form hat die nichtfibrotische HP meist einen akuten Beginn und eine besser identifizierbare Antigenquelle.
Lungenfunktionstests zeigen in der Regel restriktive Veränderungen und eine verminderte Diffusionskapazität. Die serologische Bestimmung Antigen-spezifischer IgG-Antikörper ist nicht standardisiert und korreliert inkonsistent mit der Krankheitsaktivität. Provokationstests durch Inhalation sind zwar moderat sensitiv und spezifisch, werden aber in der klinischen Praxis selten durchgeführt.
Radiologische Merkmale der HP
Die hochauflösende Computertomografie (HRCT) spielt eine entscheidende Rolle bei der Diagnose der HP und bei der Unterscheidung zwischen ihrer nichtfibrotischen und fibrotischen Form. Nichtfibrotische HP ist gekennzeichnet durch beidseitige Milchglasverschattungen, zentrilobuläre Knötchen und Mosaikperfusion mit Air Trapping, besonders gut sichtbar in exspiratorischen Aufnahmen. Ein charakteristisches Merkmal ist das „Three-Density-Sign“ – eine Mischung aus Arealen mit erhöhter, normaler und verminderter Dichte –, das hochgradig für HP spricht.
Fibrotische HP zeigt folgende Bildgebungsbefunde: Retikulation, Traktionsbronchiektasen oder Bronchiolektasen sowie „honeycombing“ (Honigwabenmuster), häufig begleitet von zentrilobulären Knötchen, Mosaikperfusion und AirTrapping, was auf einebronchioläre Obstruktion hinweist. Ein wichtiges Unterscheidungsmerkmal zur „usual interstitial pneumonia“ (UIP) bei idiopathischer pulmonaler Fibrose (IPF) ist, dass die fibrotische HP die Lungenbasen verschonen kann oder eine Prädominanz in den mittleren bis oberen Lungenfeldern aufweist. Allerdings können einige Fälle der fibrotischen HP eine bildgebende Ähnlichkeit mit UIP/IPF aufweisen, was die Diagnose erschwert. Während typische HRCT-Befunde die Diagnose HP stark unterstützen, sollten sie stets im Zusammenhang mit der klinischen Anamnese, der Expositionsbewertung und weiteren diagnostischen Untersuchungen interpretiert werden.
Pathologische Beurteilung und diagnostische Herausforderungen
Nichtfibrotische HP zeigt typischerweise eine bronchiolozentrische chronische interstitielle Entzündung, wobei das weiter entfernte Lungenparenchym oft ausgespart bleibt. Das Infiltrat besteht hauptsächlich aus Lymphozyten mit gelegentlichen Plasmazellen,wobei eine Dominanz von Plasmazellen auf eine alternative Diagnose wie „connective tissue disease-associated interstitial lung disease“ (CTD-ILD; interstitielle Lungenerkrankung bei Kollagenosen) hindeuten kann. Lymphoide Aggregate sind meist selten oder fehlen ganz, und gut ausgeprägte Keimzentren sind ungewöhnlich. Granulome oder mehrkernige Riesenzellen treten häufig im peribronchiolären Interstitium auf und können gelegentlich Cholesterinkristalle oder Schaumann-Körper enthalten. HP-Granulome weisen in der Regel nicht die feine konzentrische Fibrose auf, die für die Sarkoidose typisch ist. Fehlen Granulome, kann das histologische Muster einer zellulären nichtspezifischen interstitiellen Pneumonie (NSIP) ähneln, was die Diagnose weiter erschwert.
Fibrotische HP zeigt eine hochvariable pathologische Präsentation und gehört damit zu den diagnostisch anspruchsvollsten Formen der interstitiellen Lungenerkrankungen. Sie kann sich als peribronchioläre Fibrose, fibrotische NSIP oder sogar als UIP-ähnliches Muster präsentieren. Im Gegensatz zur UIP/IPF weist die fibrotische HP häufig eine peribronchioläre Metaplasie auf, die einen erheblichen Anteil der Bronchiolen betrifft. Während Granulome oder Riesenzellen in der fibrotischen HP seltener sind, stützt ihr Vorhandensein die Diagnose. Fibroblastenherde können gefunden werden, sind aber in der Regel weniger zahlreich als bei UIP/IPF.
Einige Differenzialdiagnosen und ihre entsprechenden pathologischen Merkmale sind in Tabelle 1 zusammengefasst.
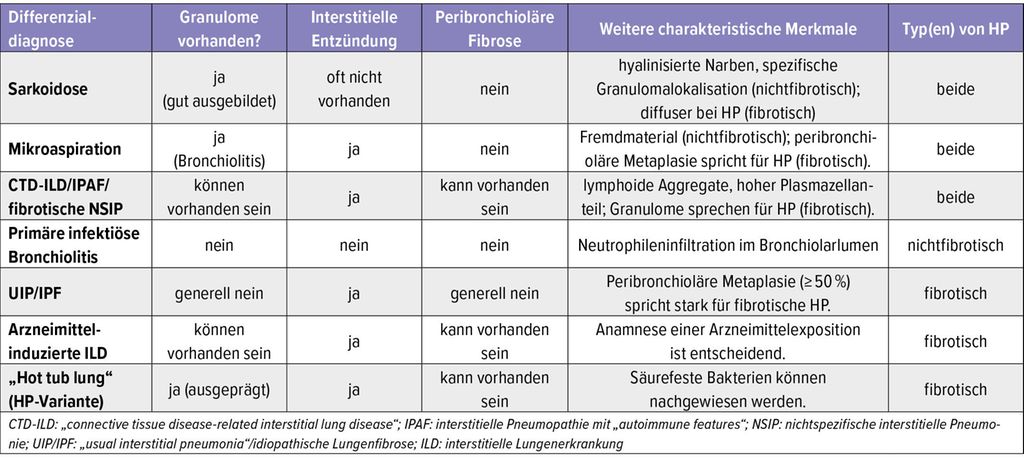
Tab. 1: Differenzialdiagnosen der Hypersensitivitätspneumonitis (HP)
Rolle der transbronchialen Biopsie und Kryobiopsie bei der Diagnose der HP
Die transbronchiale Biopsie (TBBX) kann bei der Diagnose der nichtfibrotischen HP helfen, wenn eine Kombination aus chronischer interstitieller Entzündung und dem Vorhandensein von Riesenzellen oder Granulomen nachgewiesen wird. Da eine interstitielle Entzündung allein jedoch unspezifisch ist, erfordert eine endgültige Diagnose eine Korrelation mit klinischen und radiologischen Befunden.
Die transbronchiale Kryobiopsie (TBKB) hat sich als mögliche Alternative zur chirurgischen Lungenbiopsie (CLB) etabliert, da sie größere Gewebeproben ohne Kollapsartefakte liefert. Während TBKB in einigen Studien eine gute Übereinstimmung mit CLB gezeigt hat, wurden in anderen erhebliche Unterschiede festgestellt, insbesondere bei der Unterscheidung zwischen fibrotischer HP und UIP/IPF. Eine wesentliche Einschränkung von TBKB ist die inkonsistente Detektion von Granulomen und Riesenzellen, die oft nur fleckförmig verteilt sind. Studien zeigen, dass peribronchioläre Metaplasie, ein charakteristisches Merkmal der fibrotischen HP, in Kryobiopsien zuverlässiger nachgewiesen werden kann – insbesondere wenn mehrere Proben entnommen werden. Dennoch fehlt bislang ein standardisierter Ansatz für TBKB, und die Variabilität der Gewebeprobengröße beeinflusst die diagnostische Genauigkeit. Die CLB bleibt der Goldstandard für die Diagnose der HP, insbesondere in schwierigen Fällen, in denen TBKB und andere diagnostische Verfahren keine eindeutigen Ergebnisse liefern. Obwohl CLB eine höhere diagnostische Ausbeute bietet, hat sie auch Einschränkungen – darunter Variabilität in der Probennahme sowie das Fehlen von Granulomen oder Riesenzellen in bis zu 30% der Fälle von fibrotischer HP. Trotz dieser Herausforderungen empfehlen aktuelle Leitlinien die CLB, wenn nichtinvasive Methoden wie HRCT und bronchoalveoläre Lavage keine definitive Diagnose ermöglichen.
Eine anhaltende Herausforderung bei allen Biopsieverfahren ist die Repräsentativität der gewonnenen Gewebeproben.
Bronchoalveoläre Lavage (BAL)
Die BAL spielt eine wertvolle Rolle bei der Diagnose, da sie die Immunantwort durch die Beurteilung der T-Zell-vermittelten lymphozytären Infiltration erfasst. Eine Lymphozytose ist bei nichtfibrotischer HP ausgeprägter, während die fibrotische Form oft niedrigere Werte aufweist, die invers mit dem Ausmaß der Fibrose korrelieren. Während das Fehlen einer BAL-Lymphozytose dazu beitragen kann, eine nichtfibrotische HP auszuschließen, schließt es fibrotische Formen nicht aus. Ein BAL-Lymphozytenanteil von 40 bis 60% unterstützt zwar nicht spezifisch, aber dennoch stark die Diagnose einer HP, wenn er zusammen mit klinischen und radiologischen Befunden bewertet wird. Eine BAL-Lymphozytose von über 20% kann die Diagnose insbesondere bei nichtfibrotischer HP stützen, ist jedoch nicht spezifisch, da sie auch bei anderen Erkrankungen wie der Sarkoidose vorkommen kann. Darüber hinaus hilft die BAL bei der Differenzierung der HP von anderen interstitiellen Lungenerkrankungen (ILD), wie der idiopathischen pulmonalen Fibrose (IPF), insbesondere wenn sie mit transbronchialen Biopsiebefunden kombiniert wird, wodurch die diagnostische Genauigkeit verbessert wird.
Multidisziplinäre Diagnose der HP
Die Diagnose der HP erfordert eine multidisziplinäre Diskussion unter Einbeziehungklinischer, radiologischer und pathologischer Korrelationen (Abb. 1). Eine Diagnose kann oft ohne Biopsie gestellt werden, wenn eine eindeutige Expositionsanamnese zu einem bekannten Antigen und typische HRCT-Befunde vorliegen. In Fällen mit unklarer Expositionsanamnese oder atypischen Bildgebungsmerkmalen kann jedoch eine Lungenbiopsie erforderlich sein (Abb.1). Die Interpretation von Biopsiebefunden erfordert eine sorgfältige Korrelation mit der Bildgebung. Einige Patienten mit CT-Merkmalen, die auf fibrotische HP hinweisen, können in der Biopsie ein NSIP- oder UIP-Muster aufweisen, während andere mit CT-Befunden, die mit NSIP oder UIP übereinstimmen, histologische Merkmale einer HP zeigen können. Das Fehlen eines bekannten Antigens ist mit einer schlechteren Prognose assoziiert, was die Notwendigkeit eines umfassenden diagnostischen Ansatzes unterstreicht, der alle verfügbaren klinischen, bildgebenden und histopathologischen Informationen berücksichtigt.
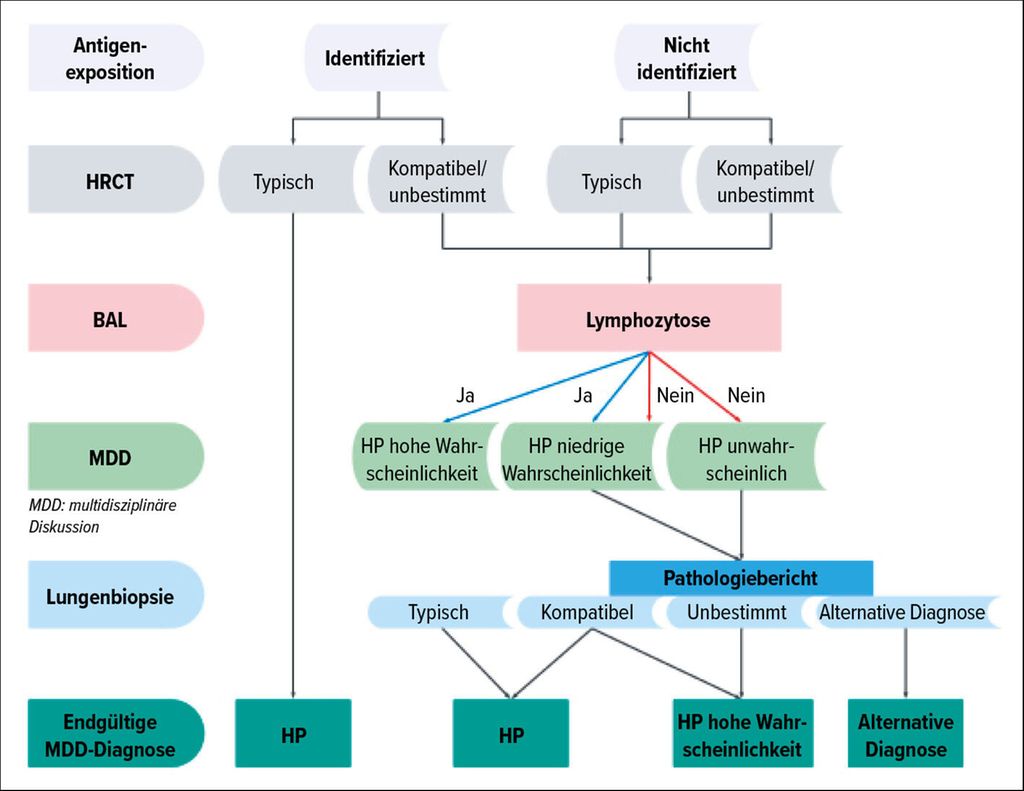
Abb. 1: Die Diagnose der HP erfordert eine multidisziplinäre Diskussion unter Einbeziehung klinischer, radiologischer und pathologischer Korrelationen
Literatur:
● Churg A: Hypersensitivity pneumonitis: new concepts andclassifications. Modern Pathol 2022; 35: 15-27 ● Marinescu DC et al.: Integration and application of clinical practice guidelines for the diagnosis of idiopathic pulmonary fibrosis and fibrotic hypersensitivity pneumonitis. Chest 2022; 162(3): 614-29 ● Moua T et al.: Challenges in the diagnosis and management of Fibrotic Hypersensitivity Pneumonitis: a practical review of current approaches. J Clin Med 2022; 11(6): 1473 ● Pérez ER et al.: Diagnosis and evaluation of hypersensitivity pneumonitis: CHEST guideline and expert panel report. Chest 2021; 160(2): e97-156 ● Yang SR et al.: Diagnosis of hypersensitivity pneumonitis: review and summary of American College of Chest Physicians statement. Am J Surg Pathol 2022; 46(4): e71-93
Das könnte Sie auch interessieren:

Allergie: wie es beginnt
Allergien sind multifaktorielle Erkrankungen, die in jedem Lebensalter, vornehmlich aber in den ersten Lebensdekaden auftreten. Auch Asthma bronchiale ist eine heterogene, ...

Asthma und der zirkadiane Rhythmus
Der zirkadiane Rhythmus spielt nicht nur beim Schlafverhalten eine bedeutende Rolle, sondern hat auch einen erheblichen Einfluss auf Asthmaanfälle und die Lungenfunktion. Die gezielte ...
